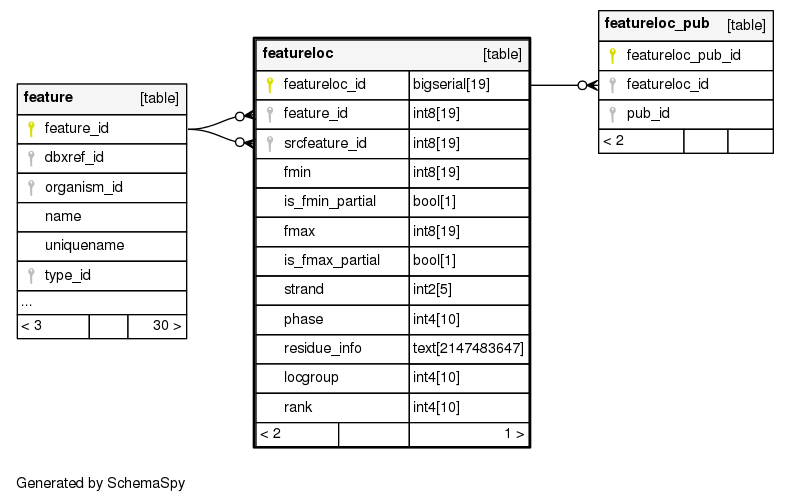
Columns
| Column | Type | Size | Foreign Key | Nullable | Default | Comments | ||
|---|---|---|---|---|---|---|---|---|
| featureloc_id | bigserial | 19 |
|
nextval('sequence.featureloc_featureloc_id_seq'::regclass) | ||||
| feature_id | int8 | 19 |
|
null | The feature that is being located. Any feature can have zero or more featurelocs. |
|||
| srcfeature_id | int8 | 19 |
|
√ | null | The source feature which this location is relative to. Every location is relative to another feature (however, this column is nullable, because the srcfeature may not be known). All locations are -proper- that is, nothing should be located relative to itself. No cycles are allowed in the featureloc graph. |
||
| fmin | int8 | 19 |
|
√ | null | The leftmost/minimal boundary in the linear range represented by the featureloc. Sometimes (e.g. in Bioperl) this is called -start- although this is confusing because it does not necessarily represent the 5-prime coordinate. Important: This is space-based (interbase) coordinates, counting from zero. To convert this to the leftmost position in a base-oriented system (eg GFF, Bioperl), add 1 to fmin. |
||
| is_fmin_partial | bool | 1 |
|
false | This is typically false, but may be true if the value for column:fmin is inaccurate or the leftmost part of the range is unknown/unbounded. |
|||
| fmax | int8 | 19 |
|
√ | null | The rightmost/maximal boundary in the linear range represented by the featureloc. Sometimes (e.g. in bioperl) this is called -end- although this is confusing because it does not necessarily represent the 3-prime coordinate. Important: This is space-based (interbase) coordinates, counting from zero. No conversion is required to go from fmax to the rightmost coordinate in a base-oriented system that counts from 1 (e.g. GFF, Bioperl). |
||
| is_fmax_partial | bool | 1 |
|
false | This is typically false, but may be true if the value for column:fmax is inaccurate or the rightmost part of the range is unknown/unbounded. |
|||
| strand | int2 | 5 |
|
√ | null | The orientation/directionality of the location. Should be 0, -1 or +1. |
||
| phase | int4 | 10 |
|
√ | null | Phase of translation with respect to srcfeature_id. Values are 0, 1, 2. It may not be possible to manifest this column for some features such as exons, because the phase is dependant on the spliceform (the same exon can appear in multiple spliceforms). This column is mostly useful for predicted exons and CDSs. |
||
| residue_info | text | 2147483647 |
|
√ | null | Alternative residues, when these differ from feature.residues. For instance, a SNP feature located on a wild and mutant protein would have different alternative residues. for alignment/similarity features, the alternative residues is used to represent the alignment string (CIGAR format). Note on variation features; even if we do not want to instantiate a mutant chromosome/contig feature, we can still represent a SNP etc with 2 locations, one (rank 0) on the genome, the other (rank 1) would have most fields null, except for alternative residues. |
||
| locgroup | int4 | 10 |
|
0 | This is used to manifest redundant, derivable extra locations for a feature. The default locgroup=0 is used for the DIRECT location of a feature. Important: most Chado users may never use featurelocs WITH logroup > 0. Transitively derived locations are indicated with locgroup > 0. For example, the position of an exon on a BAC and in global chromosome coordinates. This column is used to differentiate these groupings of locations. The default locgroup 0 is used for the main or primary location, from which the others can be derived via coordinate transformations. Another example of redundant locations is storing ORF coordinates relative to both transcript and genome. Redundant locations open the possibility of the database getting into inconsistent states; this schema gives us the flexibility of both warehouse instantiations with redundant locations (easier for querying) and management instantiations with no redundant locations. An example of using both locgroup and rank: imagine a feature indicating a conserved region between the chromosomes of two different species. We may want to keep redundant locations on both contigs and chromosomes. We would thus have 4 locations for the single conserved region feature - two distinct locgroups (contig level and chromosome level) and two distinct ranks (for the two species). |
|||
| rank | int4 | 10 |
|
0 | Used when a feature has >1 location, otherwise the default rank 0 is used. Some features (e.g. blast hits and HSPs) have two locations - one on the query and one on the subject. Rank is used to differentiate these. Rank=0 is always used for the query, Rank=1 for the subject. For multiple alignments, assignment of rank is arbitrary. Rank is also used for sequence_variant features, such as SNPs. Rank=0 indicates the wildtype (or baseline) feature, Rank=1 indicates the mutant (or compared) feature. |
Table contained -1 rows
Indexes
| Constraint Name | Type | Sort | Column(s) |
|---|---|---|---|
| featureloc_pkey | Primary key | Asc | featureloc_id |
| featureloc_c1 | Must be unique | Asc/Asc/Asc | feature_id + locgroup + rank |
| featureloc_idx1 | Performance | Asc | feature_id |
| featureloc_idx2 | Performance | Asc | srcfeature_id |
| featureloc_idx3 | Performance | Asc/Asc/Asc | srcfeature_id + fmin + fmax |
Relationships
The following entity relationship (ER) diagram shows the relationships involving the featureloc table.
Show relationships within degrees of separation